Beta-Thalassämien zählen zu den häufigsten monogenetischen hereditären Erkrankungen und sind vor allem in Ländern des Mittelmeerraumes, des Nahen und Mittleren Ostens sowie auf dem indischen Subkontinent, in Südostasien und in Afrika beheimatet. Da Menschen mit der heterozygoten Beta-Thalassämie vor schweren Verläufen der durch den Erreger Plasmodium falciparum ausgelöstem Malaria tropica geschützt sind, hat die natürliche Auslese eine Häufung der genetischen Variante in Malaria-Regionen begünstigt.
Die Prävalenz des Trägerstatus liegt dort zwischen 5 und 30 % , wohingegen die Erkrankung in Deutschland vergleichsweise selten auftritt: Die Prävalenz der heterozygoten Beta-Thalassämie in der aus Deutschland stammenden Bevölkerung wird auf 0,01 % geschätzt. Aufgrund von Migration konnte in den letzten Jahrzenten allerdings eine erhebliche Zunahme von Patient:innen und Trägern im mitteleuropäischen Raum verzeichnet werden.
Da die Vererbung autosomal erfolgt, sind Männer genauso häufig betroffen wie Frauen.
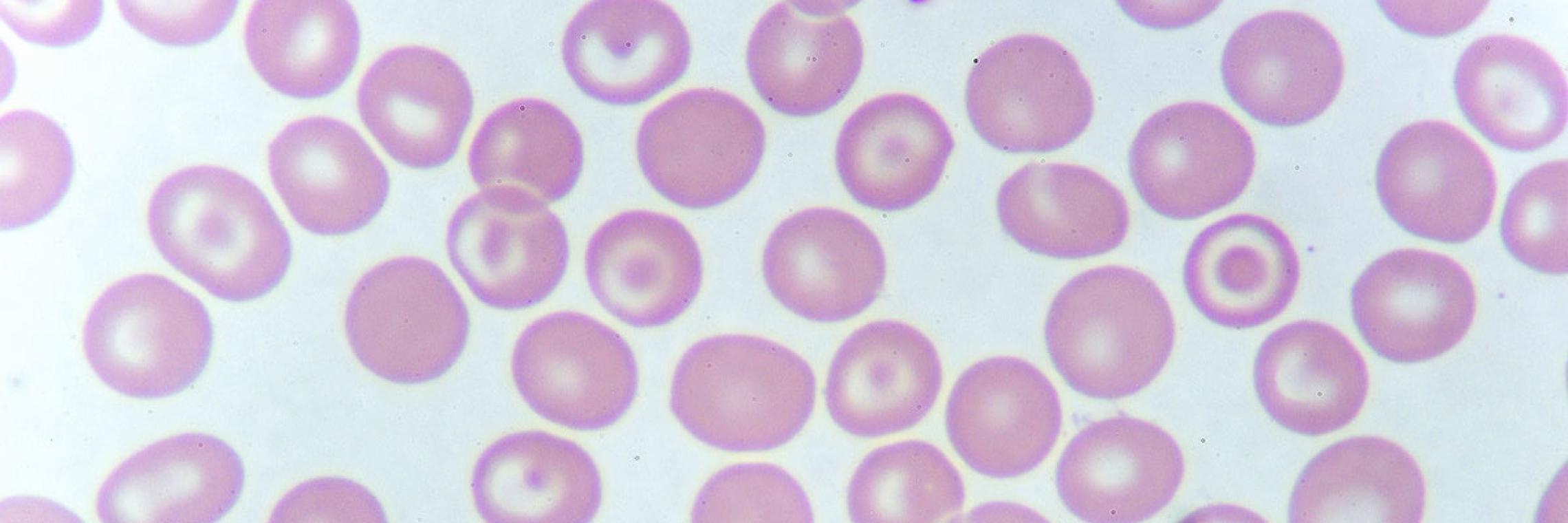
Die Thalassaemia major verursacht hochgradige Anämie, die bei betroffenen Kindern ohne Behandlung noch vor dem Alter von 3 Jahren zum Tod führt. Eine höhere Lebenserwartung kann allerdings durch regelmäßige Bluttransfusionen und Chelattherapien oder allogene Bluttransplantationen erzielt werden. Auch die Thalassaemia intermedia erfordert regelmäßige klinische Kontrollen, um einen möglichen Transfusionsbedarf rechtzeitig zu erkennen und die Transfusionstherapie analog zum Vorgehen bei der Thalassaemia major zu beginnen.