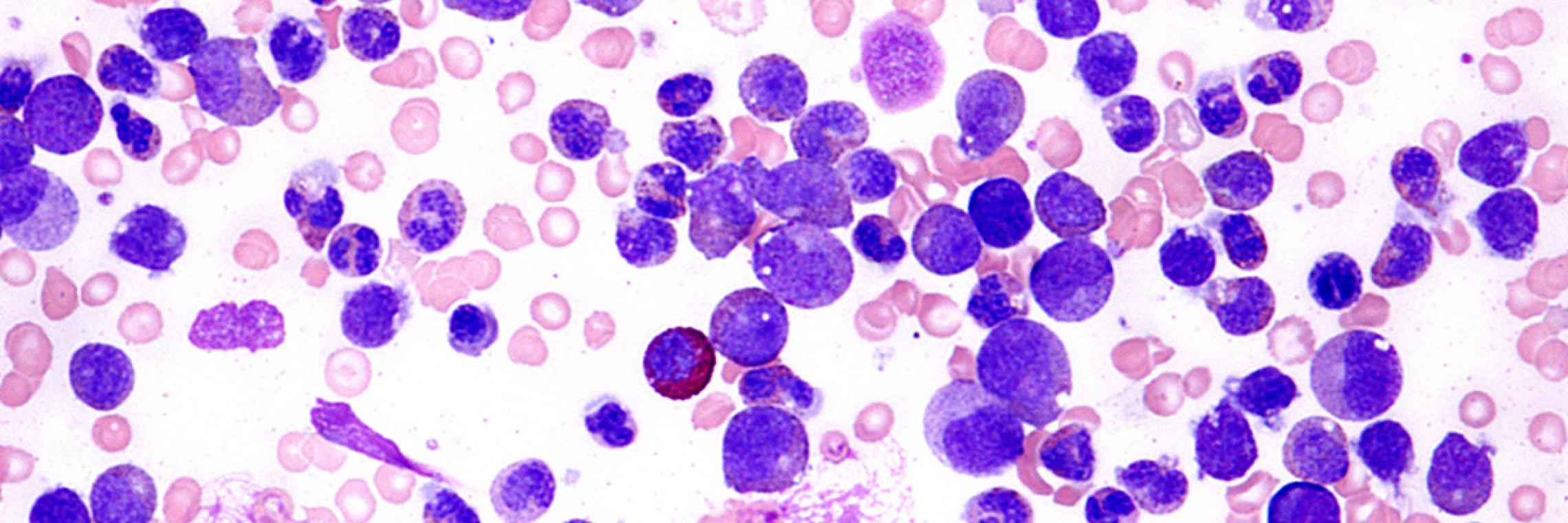




Das Plasmozytom oder Multiple Myelom ist eine maligne hämatologische Erkrankung, die durch die zumeist fortschreitende, unkontrollierte Proliferation von malignen Plasmazellen (Myelomzellen) im Knochenmark und durch Produktion von pathologischem monoklonalem Immunglobulin, dem sogenannten M-Protein, charakterisiert ist. Die Erkrankung wird auch als Morbus Kahler bezeichnet.Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie e. V. (DGHO), Onkopedia. Multiples Myelom Leitlinie. Stand Mai 2018. https://www.onkopedia.com/de/onkopedia/guidelines/multiples-myelom/@@guideline/html/index.html; abgerufen am 10.03.2020. Possinger K, Eucker J, Regierer AC. Facharztwissen Hämatologie Onkologie. München: Urban & Fischer in Elsevier, 2017.