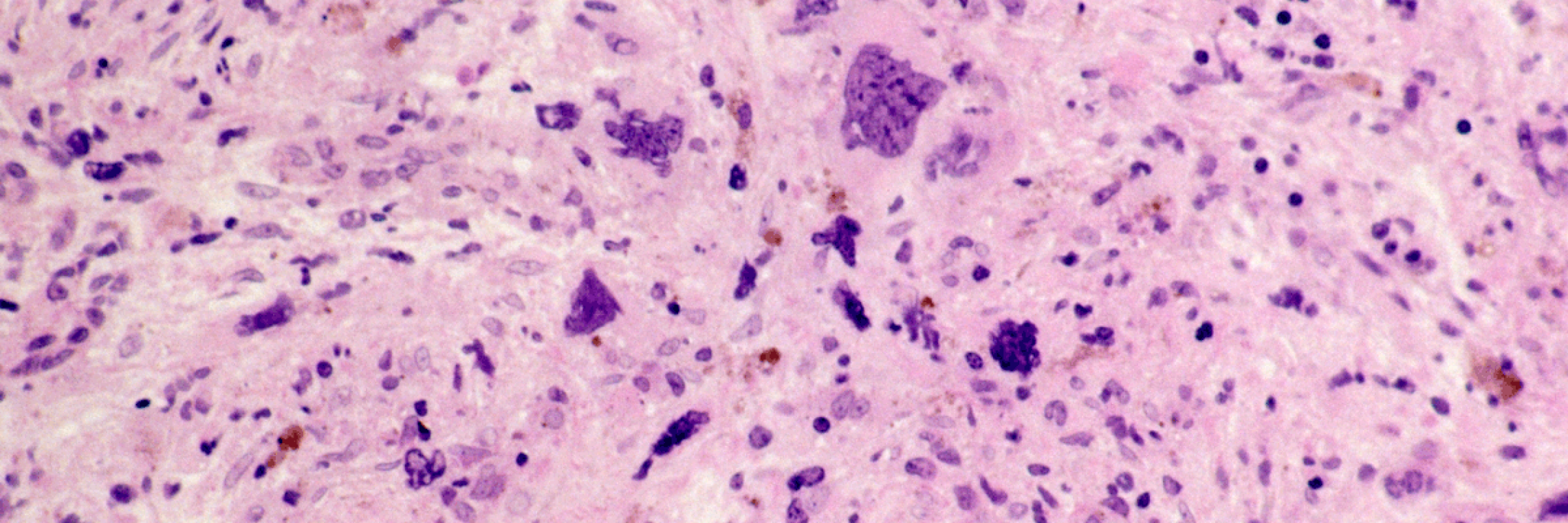




Die Myelofibrose (MF) ist eine seltene hämatologische Erkrankung und gehört zu den chronischen myeloproliferativen Neoplasien. Man unterscheidet zwischen einer Vorstufe der präfibrotischen primären Myelofibrose (präPMF) mit Hyperproliferation einer oder mehrere hämatopoetischer Zelllinien, einer primären Myelofibrose (PMF) mit zunehmender Fibrosierung des Knochenmarks und sekundären Formen, welche sich aus einer Polycythaemia vera (Post-PV-Myelofibrose) oder einer essenziellen Thrombozythämie (Post-ET-Myelofibrose) entwickeln können. Männer erkranken etwas häufiger als Frauen. Die Prognose wird von klinischen und genetischen Parametern bestimmt und kann mithilfe von verschiedenen Scores berechnet werden.Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie e. V. (DGHO). Onkopedia Leitlinie Primäre Myelofibrose (PMF). Stand Dezember 2018. Abgerufen am 20.05.2021. https://www.onkopedia.com/de/onkopedia/guidelines/primaere-myelofibrose-pmf/@@guideline/html/index.html#litID0EJ4AG Die Myelofibrose birgt das Risiko des Übergangs in eine akute myeloische Leukämie (AML), weitere wichtige Komplikationen stellen Infektionen und kardiovaskuläre Erkrankungen dar.Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie e. V. (DGHO). Onkopedia Leitlinie Primäre Myelofibrose (PMF). Stand Dezember 2018. Abgerufen am 20.05.2021. https://www.onkopedia.com/de/onkopedia/guidelines/primaere-myelofibrose-pmf/@@guideline/html/index.html#litID0EJ4AG Frederiksen H, Szepligeti S, Bak M, Ghanima W, Hasselbalch HC, Christiansen CF. Vascular Diseases In Patients With Chronic Myeloproliferative Neoplasms - Impact Of Comorbidity. Clinical epidemiology. 2019;11:955-967. doi:10.2147/CLEP.S216787 Barbui T, Carobbio A, Cervantes F, et al. Thrombosis in primary myelofibrosis: incidence and risk factors. Blood. 2010;115(4):778-82. doi:10.1182/blood-2009-08-238956